Recently, a major breakthrough in elementary reaction dynamics was made by a DICP team leading by Prof. Xueming Yang and Prof. Donghui Zhang.
In many chemical reactions that play important roles in combustion chemistry, atmosphere chemistry and astrochemistry, a significant portion of reactant molecules are populated in vibrationally excited levels and molecular vibration is a strong thrust to overcome the reaction barrier. But little is known about how the vibrational excitation in the reacting molecules change the reactive process.
The reaction of fluorine atom with hydrogen molecule is one of the most important benchmark systems in the history of molecular reaction dynamics. In the past few years, this reaction was intensively explored by the DICP team. Dynamical resonances, the breakdown of Born–Oppenheimer approximation and partial wave resonances were observed in this reaction, and the mechanism were elucidated. In these works, the hydrogen molecule was populated in the vibrationally ground state. So a interesting question is: if the reacting hydrogen molecule is vibrationally excited, how will this benchmark reaction change?
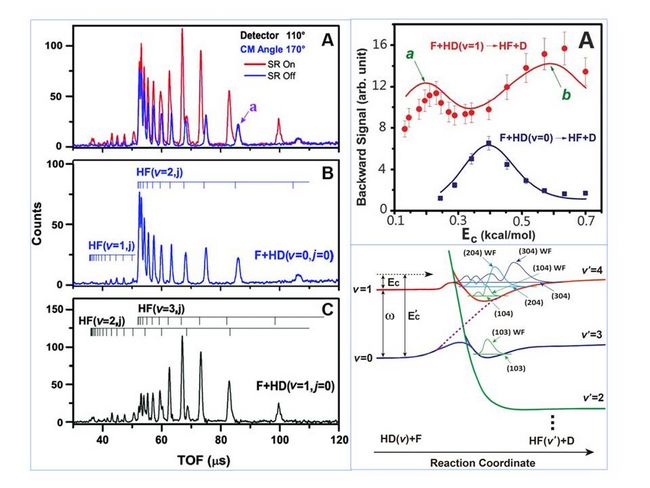
In order to answer this question, the team built a single-longitudinal-mode optical parametrical oscillator system, and developed a highly efficient technique for preparation of an intense beam of vibrationally excited hydrogen molecules (J. Phys. Chem. Lett. 2013, 4, 368-371). Based on this technique, a high-resolution crossed-molecular-beam experiment on the vibrationally excited HD (v = 1) + F → HF + D reaction was successfully conducted at DICP.
Two broad peaks for backward-scattered HF (v= 2 & 3) products clearly emerge at collision energies of 0.21 kcal/mol and 0.62 kcal/mol from differential cross sections measured over a range of energies. These features are attributed to excited Feshbach resonances trapped in the peculiar HF (v = 4)–D vibrationally adiabatic potential in the post-barrier region. Quantum dynamics calculations on a highly accurate potential energy surface show that these resonance states correlate to the HD (v= 1) state in the entrance channel and therefore can only be accessed by the vibrationally excited HD reagent.
This work clearly reveals that initial vibrational excitation not only provides energy required for the reaction but also gives rise to a proper adiabatic pathway that accesses the dynamical resonances in the high-energy region, whereas translational energy cannot provide such a pathway. The results show that vibrationally excited molecular reactions provide a unique context for probing dynamical resonances in chemical reactions when the vibrationally adiabatic potential, which correlates to the vibrationally excited state of the reagent in the entrance channel, supports quasibound resonance states.
This work was supported by the National Natural Science Foundation of China, the Ministry of Science and Technology of China, and the Chinese Academy of Sciences. The paper was published in Science Magazine (Science , 342, 1499).